How Generative AI Transforms Pharmaceutical Data Management Solutions
Table Of Content
Published Date :
11 Mar 2026
- Data volume in US pharma is rising faster than legacy systems can handle.
- Generative AI in pharmaceutical industry reduces manual drafting and speeds up data workflows.
- Biggest impact areas include regulatory submissions, clinical reconciliation, safety case processing, and batch documentation review.
- Efficiency gains of 15 to 30 percent can improve timelines and reduce operational costs.
- Strong governance and human validation keep compliance intact.
- Intelligent integration strengthens existing data management solutions without replacing core systems.
US pharmaceutical companies are generating more data than ever before. Pharmaceutical data management solutions are under increasing pressure as data volumes grow across clinical, regulatory, and manufacturing workflows. Clinical trials across multiple sites, post-market surveillance reports, manufacturing batch logs, and real-world evidence streams are expanding at a pace that legacy systems were never built to handle. At the same time, regulatory oversight from FDA, HIPAA, and GxP frameworks continues to tighten, leaving little room for manual errors or delayed documentation.
Here’s the challenge. Most organizations still rely on fragmented platforms and semi-manual workflows to manage critical information. That fragmentation slows submissions, increases compliance exposure, and quietly inflates operational costs.
This is where generative AI in pharmaceutical industry is starting to reshape strategy. Not as a buzzword, but as a practical layer that enhances how data is captured, structured, reviewed, and governed. For leadership teams focused on speed, compliance, and scalability, the conversation is no longer optional. It is strategic.
What are Pharmaceutical Data Management Solutions
Pharmaceutical data management solutions refer to systems and platforms used to collect, validate, store, and analyze data across clinical trials, regulatory processes, pharmacovigilance, and manufacturing operations. These solutions form the backbone of modern drug development and commercialization. They control how information is captured, validated, stored, governed, and analyzed across clinical, regulatory, safety, and manufacturing functions. When these systems operate in sync, decision-making becomes faster and risk exposure drops significantly.
In most US pharma environments, data flows through platforms such as:
- Electronic Data Capture systems for clinical trials
- Laboratory Information Management Systems for research and testing
- Clinical Trial Management Systems for site coordination
- ERP platforms for supply chain and finance
- Pharmacovigilance databases for adverse event tracking
But here’s where friction shows up. Data often lives in silos. Reconciliation between trial sites may take weeks. Audit trails may require manual compilation. And when leadership requests a consolidated performance snapshot, teams scramble across systems.
Poor visibility doesn’t just slow operations. It delays submissions, increases review cycles, and can trigger costly remediation efforts during inspections. In an industry where timelines directly influence revenue, disconnected data architecture becomes more than an IT issue. It becomes a strategic liability.
Is Your Pharma Data Infrastructure Ready for AI?
Discover how AI can streamline regulatory documentation, improve data visibility, and strengthen compliance across pharmaceutical data management workflows.
Key Challenges & Solution in Pharmaceutical Data Management
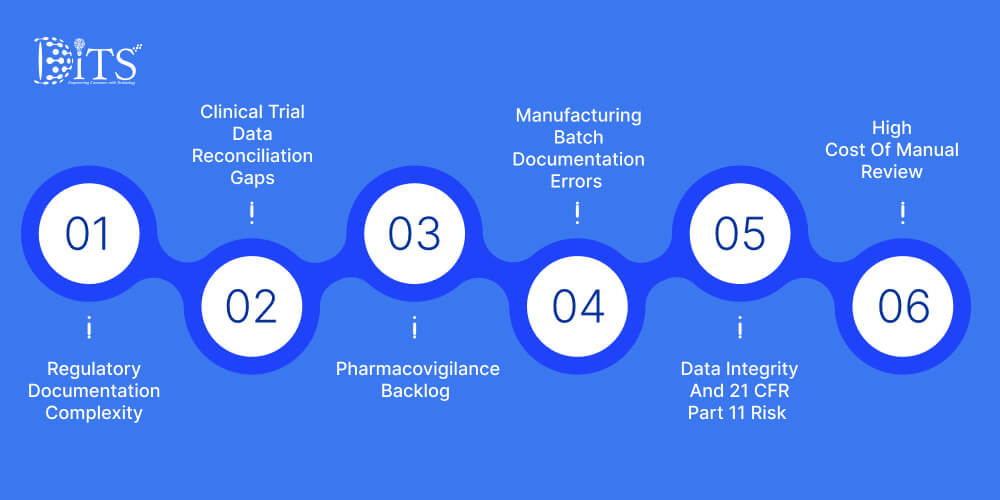
Rapid data growth has exposed structural weaknesses across many US pharma organizations. As operations expand, inefficiencies that once felt manageable now carry measurable financial and regulatory consequences. Below are the pressure zones leadership teams confronting most often.
Regulatory Documentation Complexity
IND, NDA, and BLA submissions demand extreme precision. Clinical summaries must align with statistical outputs, safety data must reconcile across datasets, and formatting must meet U.S. Food and Drug Administration expectations. Even minor discrepancies can trigger information requests that delay approvals by weeks or months. Regulatory teams frequently dedicate hundreds of hours to cross-checking documents manually, driving up submission costs.
Solution
Pharmaceutical companies are addressing this challenge by implementing AI-enabled regulatory document management platforms. These systems automatically map clinical datasets, statistical reports, and safety summaries into standardized submission structures. Built-in validation checks identify inconsistencies early and ensure alignment with submission requirements. As a result, regulatory teams reduce manual review time, accelerate document preparation cycles, and improve submission accuracy.
Clinical Trial Data Reconciliation Gaps
Multi-site trials introduce variability. Different data entry habits, inconsistent coding standards, and late protocol deviation reporting create reconciliation bottlenecks. Data managers often spend significant time aligning datasets before locking trials, which directly affects submission readiness and investor confidence.
Solution
Unified clinical data platforms help consolidate information from multiple trial sites into a centralized environment. Standardized data models, automated validation rules, and real-time monitoring dashboards ensure consistency across datasets. By identifying discrepancies earlier in the trial lifecycle, organizations can reduce reconciliation delays and ensure that trial databases are ready for regulatory submission more quickly.
Pharmacovigilance Backlog
Adverse event reporting volumes continue to increase. Case intake, narrative drafting, medical review, and signal detection remain labor-heavy processes. When case backlogs grow, compliance risk increases—and regulators do not tolerate reporting delays.
Solution
AI-driven pharmacovigilance systems automate many safety monitoring processes. Natural language processing tools can extract key details from adverse event reports, generate initial case narratives, and prioritize cases based on severity. Automated workflows also help safety teams maintain consistent reporting timelines, reducing backlog risk while maintaining regulatory compliance.
Manufacturing Batch Documentation Errors
Electronic batch records still require detailed review cycles. A missing entry or improperly logged deviation can delay product release, impacting revenue forecasts and supply commitments. Nobody wants production lines paused because of documentation gaps.
Solution
Modern manufacturing execution systems with electronic batch record automation help eliminate manual documentation gaps. Real-time validation rules ensure that operators complete required fields before advancing production steps. Digital deviation tracking and automated approval workflows also streamline quality review processes, allowing products to move through release cycles more efficiently.
Data Integrity And 21 CFR Part 11 Risk
Audit trails must be complete, tamper-proof, and transparent. Manual handling increases the likelihood of errors and incomplete logs. Remediation efforts following inspection findings often cost far more than proactive system strengthening.
Solution
Pharmaceutical organizations are strengthening compliance with 21 CFR Part 11 by implementing secure data architectures that support encrypted audit trails, role-based access controls, and automated compliance monitoring. These systems capture every data change and maintain immutable records, ensuring transparency during regulatory inspections and reducing the risk of costly compliance remediation.
High Cost Of Manual Review
Across regulatory affairs, clinical operations, safety monitoring, and manufacturing, manual review remains one of the largest hidden expenses. When documentation cycles stretch, payroll and compliance exposure increase simultaneously.
Solution
AI-powered document review and workflow automation tools significantly reduce the burden of manual validation. These systems can analyze large volumes of documentation, detect inconsistencies, and flag missing information before final review stages. By automating repetitive checks, organizations reduce operational costs, accelerate documentation cycles, and improve overall efficiency across departments.
How Generative AI in Pharmaceutical Industry Improves Data Management
When applied strategically, generative AI in pharmaceutical industry does not replace core systems. It strengthens them. It reduces repetitive cognitive workload, accelerates documentation cycles, and enhances consistency across regulated environments. The real value shows up in specific workflows where time, accuracy, and compliance intersect.
Intelligent Data Extraction and Structuring
Clinical notes, investigator comments, lab observations, and monitoring reports often exist in free-text form. Converting that information into structured datasets traditionally requires manual abstraction.
Generative models can:
- Extract key clinical variables from unstructured narratives
- Standardize terminology across sites
- Automatically tag metadata for faster retrieval
What once took weeks of manual review can be shortened significantly. Data becomes searchable, analyzable, and submission-ready earlier in the cycle.
Automated Regulatory Document Drafting
Regulatory teams spend extensive time compiling submission modules. Generative systems can assist by drafting structured sections based on validated source data, reducing first-level writing workload.
Applications include:
- Drafting clinical overviews from trial datasets
- Summarizing safety findings
- Formatting content to match submission templates
Human review remains critical, but preparation time drops. That acceleration directly impacts FDA submission timelines.
Clinical Trial Data Harmonization
Site variability introduces inconsistency. Generative systems can flag anomalies, detect mismatched coding patterns, and identify protocol deviations earlier in the process.
Instead of discovering discrepancies during final reconciliation, teams receive proactive alerts. That shift shortens database lock timelines and strengthens trial integrity.
Pharmacovigilance Acceleration
Safety teams manage growing case volumes. Generative AI assists by summarizing adverse event reports, extracting relevant patient history, and preparing narrative drafts for medical review.
Signal detection also improves when structured summaries feed analytics engines faster. Case processing time decreases while maintaining oversight controls.
Manufacturing and Batch Record Optimization
Electronic batch records generate extensive documentation. Intelligent review systems can identify missing entries, flag inconsistencies, and detect deviation patterns before lot release.
This early validation reduces rework and protects supply continuity. And in manufacturing, continuity directly translates into revenue stability.
Knowledge Management Across R&D
Large pharmaceutical organizations accumulate decades of research data. Yet locating historical trial findings or previous regulatory correspondence often requires manual searching across repositories.
Generative tools enable enterprise-level semantic search, allowing teams to retrieve context-rich information within minutes instead of hours. Research cycles tighten. Institutional knowledge becomes accessible instead of buried.
When embedded carefully within pharmaceutical data management solutions, this intelligence layer transforms documentation-heavy operations into more agile, insight-driven processes.
Real-World Use Case: Generative AI in Pharmaceutical Data Management
A mid-sized US pharmaceutical company managing multi-site clinical trials faced consistent delays in regulatory submissions due to fragmented data systems and manual documentation processes.
Challenge
- Clinical data inconsistencies across multiple trial sites
- High manual effort in regulatory document preparation
- Delays in submission timelines impacting product launch strategy
Solution
The organization implemented a generative AI layer integrated with its existing pharmaceutical data management solutions.
Key capabilities included:
- Automated extraction and structuring of clinical and safety data
- AI-assisted drafting of regulatory documents and summaries
- Real-time detection of discrepancies across datasets
Results
- 28% reduction in regulatory document preparation time
- 22% faster clinical database lock timelines
- Significant reduction in manual reconciliation workload
Business Impact
The company improved submission predictability, reduced operational overhead, and strengthened compliance readiness. This demonstrates how generative AI in pharmaceutical industry can deliver measurable improvements without replacing validated systems.
How to Choose the Right Pharmaceutical Data Management Solutions with AI
Selecting the right pharmaceutical data management solutions is a strategic decision that directly impacts regulatory timelines, compliance risk, and operational efficiency. When generative AI is involved, the evaluation must go beyond features and focus on long-term scalability and regulatory alignment.
Build vs Buy Approach
Custom-Built AI Solutions: Suitable for large pharmaceutical organizations with complex workflows and internal engineering capabilities. These solutions offer full control, deeper customization, and better alignment with proprietary processes, but require higher upfront investment and longer implementation timelines.
Pre-built Platforms with AI Capabilities: Faster to deploy and easier to scale initially. These solutions reduce development effort but may have limitations in customization, integration flexibility, and regulatory adaptability.
Key Evaluation Criteria
When assessing pharmaceutical data management solutions with AI, decision-makers should consider:
- Compliance readiness (21 CFR Part 11, GxP validation support)
- Integration capability with EDC, LIMS, CTMS, ERP, and pharmacovigilance systems
- Data governance, audit trails, and traceability
- AI explainability and human-in-loop validation support
- Scalability across clinical, regulatory, and manufacturing workflows
Vendor Selection Considerations
- Proven experience in the pharmaceutical industry
- Demonstrated use cases in regulatory, clinical, or safety workflows
- Ability to support validation documentation and audit readiness
- Long-term support, upgrades, and scalability roadmap
Organizations that align solution selection with compliance requirements and operational priorities reduce implementation risk while accelerating ROI from generative AI in pharmaceutical industry.
Cost and ROI Impact for US Pharmaceutical Companies
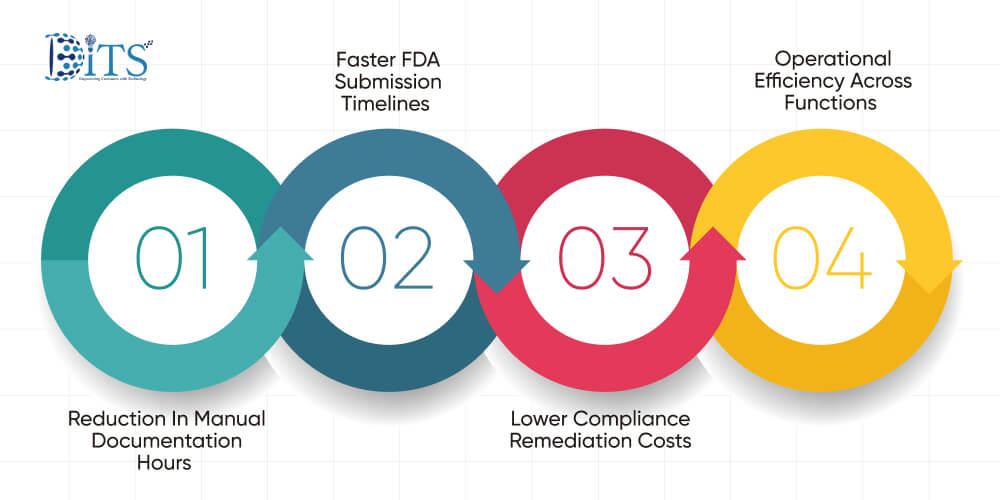
Executive teams rarely invest in technology for novelty. They invest for measurable return. When generative intelligence is embedded within pharmaceutical data management solutions, financial impact becomes visible across several cost centers.
Reduction In Manual Documentation Hours
Regulatory affairs teams often dedicate thousands of staff hours annually to document compilation and cross-verification. Even a 20 to 30 percent reduction in drafting workload can translate into substantial payroll savings. In large submission programs, that may represent hundreds of thousands of dollars per cycle.
Faster FDA Submission Timelines
Accelerating submission readiness by even four to six weeks can influence market entry strategy. For products with projected annual revenues exceeding 200 million dollars, earlier approval materially affects revenue realization.
Lower Compliance Remediation Costs
FDA observations related to documentation gaps or incomplete audit trails frequently trigger corrective action programs. Remediation efforts can cost from 250,000 to several million dollars depending on scope. Proactive automation reduces likelihood of such findings.
Operational Efficiency Across Functions
Clinical operations, safety monitoring, and manufacturing documentation benefit from shorter review cycles. When database locks happen earlier and batch releases avoid rework, operational stability improves.
Below is a simplified impact view:
| Area | Potential Efficiency Gain | Financial Effect |
| Regulatory Drafting | 20% to 30% reduction in preparation time | Lower labor cost per submission and reduced outsourcing expenses |
| Clinical Data Reconciliation | 15% to 25% faster database lock | Earlier submission readiness and improved timeline predictability |
| Pharmacovigilance Processing | Approximately 25% acceleration in case handling | Reduced backlog, lower overtime cost, and improved compliance posture |
| Batch Record Review | 10% to 20% reduction in documentation errors | Fewer production delays and minimized rework costs |
Estimated Optimization Zones
Cost efficiencies typically emerge in:
- Clinical operations budgets
- Pharmacovigilance staffing
- Regulatory documentation outsourcing
- Manufacturing compliance review
Return is not just cost avoidance. It is risk mitigation and timeline compression. And in pharmaceutical markets, time carries significant financial weight.
Cost of Pharmaceutical Data Management Solutions with AI
The cost of implementing pharmaceutical data management solutions integrated with generative AI depends on system complexity, integration scope, and regulatory requirements. For US pharmaceutical companies, investment typically spans multiple cost components. The total cost depends on whether the organization adopts a custom-built solution, integrates AI into existing validated systems, or licenses a platform with prebuilt capabilities.
Key Cost Components
1. Development or Licensing Cost
- Custom AI development: $80,000 to $300,000+ depending on scope
- Pre-built platforms: Subscription or license-based pricing models
2. Integration Cost
- Connecting AI with EDC, LIMS, CTMS, ERP, and pharmacovigilance systems
- API development, middleware layers, and validation workflows
3. Data Preparation and Model Training
- Data cleaning, structuring, and annotation
- Model fine-tuning for pharmaceutical-specific use cases
4. Compliance and Validation Cost
- GxP validation documentation
- 21 CFR Part 11 compliance setup
- Audit trail and testing frameworks
5. Ongoing Maintenance and Scaling
- Model monitoring and updates
- Cloud infrastructure and storage costs
- Continuous compliance monitoring
ROI Timeline
Most pharmaceutical organizations begin to see measurable ROI within 3 to 6 months when AI is applied to high-impact workflows such as:
- Regulatory document drafting
- Pharmacovigilance case processing
- Clinical data reconciliation
Why Choose DITS For Generative AI Data Management Software
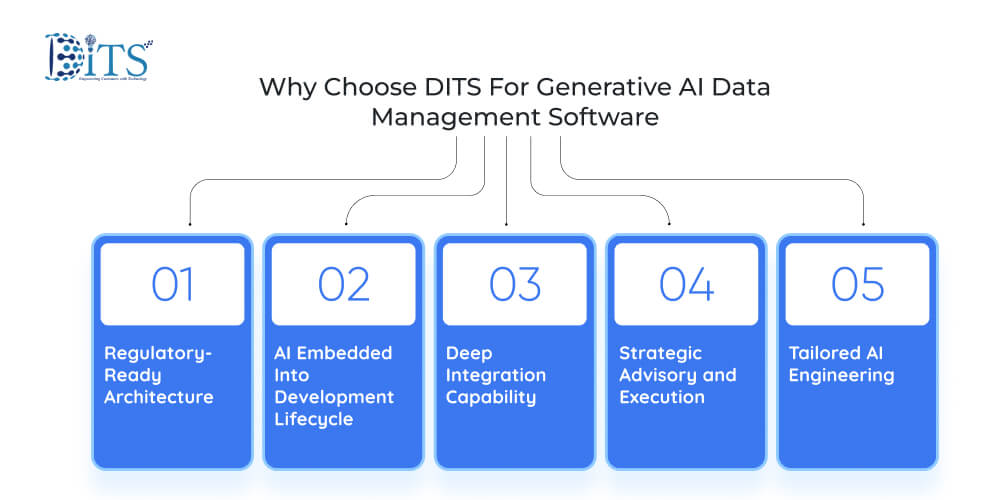
Selecting a technology partner in regulated pharmaceutical environment requires more than technical capability. It demands regulatory awareness, engineering discipline, and long-term scalability thinking. That is where DITS differentiates.
Regulatory-Ready Architecture
We design and deploy intelligence layers that align with 21 CFR Part 11, GxP documentation controls, and structured validation workflows. Systems are built with auditability, traceability, and role-based governance embedded from day one. Compliance is not treated as an afterthought. It is foundational.
AI Embedded Into Development Lifecycle
At DITS, AI is not only delivered to clients. It is integrated into how we build software. We use intelligent systems for code generation assistance, quality assurance automation, code quality monitoring, and platform customization. This strengthens delivery speed while maintaining structured validation discipline.
Deep Integration Capability
Pharma ecosystems are complex. EDC, LIMS, CTMS, ERP, and pharmacovigilance platforms must remain stable. Through specialized AI integration services, we embed intelligence within validated infrastructures rather than replacing them. This minimizes disruption and protects operational continuity.
Strategic Advisory and Execution
Technology alone does not drive transformation. Our digital transformation consulting approach evaluates architecture maturity, identifies high-impact use cases, and designs phased deployment strategies aligned with executive priorities. Roadmaps are practical, measurable, and risk-aware.
Tailored AI Engineering
We provide enterprise-grade generative AI development services designed specifically for pharmaceutical workflows such as regulatory drafting, safety narrative support, structured data extraction, and manufacturing documentation review. Each deployment is customized to operational context, not delivered as a generic template.
When intelligence is deployed in regulated industries, precision matters. DITS combines engineering rigor, compliance alignment, and operational insight to ensure generative AI in pharmaceutical industry strengthens business performance rather than introducing uncertainty.
Looking to Reduce Manual Documentation Effort?
Learn how intelligent automation can streamline clinical data processing, regulatory drafting, and safety reporting without disrupting existing pharmaceutical systems.
Conclusion
Pharmaceutical organizations are struggling with how to manage it efficiently, securely, and in a way that accelerates regulatory progress rather than slowing it down. Clinical expansion, safety reporting growth, and manufacturing scale have exposed limits of traditional systems.
Generative AI in pharmaceutical industry introduces a practical shift. It reduces repetitive documentation workload, strengthens structured data visibility, and supports compliance discipline without dismantling validated infrastructure. When implemented with governance and human oversight, it becomes an operational advantage rather than a regulatory risk.
For executive teams, decision is not whether intelligence will influence pharmaceutical data management solutions. It already is. The real decision centers on how early to adopt structured augmentation that protects compliance while improving speed and predictability.
Organizations that move thoughtfully can compress timelines, reduce review costs, and improve inspection readiness. Those that delay may find manual complexity growing faster than control frameworks can contain.
Frequently Asked Questions
What are pharmaceutical data management solutions used for?
Pharmaceutical data management solutions are used to manage, validate, and analyze data across clinical trials, regulatory submissions, pharmacovigilance, and manufacturing processes. They help ensure compliance, improve data accuracy, and accelerate decision-making across the drug development lifecycle.
Is generative AI safe to use in regulated pharmaceutical environments?
Yes, when deployed with proper validation controls, audit trails, and human review checkpoints. In regulated settings, AI systems must operate within structured governance frameworks that align with 21 CFR Part 11 and GxP requirements. The key is disciplined implementation rather than experimentation without oversight.
How does generative AI improve pharmaceutical data management solutions?
It reduces repetitive documentation workload, accelerates regulatory drafting, structures unorganized clinical data, and enhances visibility across systems. Instead of replacing validated platforms, it strengthens them by adding contextual intelligence and reducing manual reconciliation cycles.
Can DITS Generative AI Development Services customize solutions for regulatory workflows?
Yes. DITS Generative AI Development Services for pharmaceutical data management are designed around specific operational needs such as IND drafting support, pharmacovigilance narrative preparation, structured data extraction, and batch documentation review. Each deployment is engineered with compliance, traceability, and integration discipline built into the architecture.
Will AI replace regulatory or safety professionals?
No. AI systems reduce first-level drafting and data preparation effort, but subject matter experts remain responsible for review, validation, and final approval. The objective is to improve efficiency and reduce cognitive overload, not remove accountability.
How does DITS ensure compliance when integrating generative AI into pharma systems?
DITS combines Generative AI Development Services for pharmaceutical data management with validated engineering practices. Solutions are embedded within existing infrastructures through controlled integration layers, supported by documentation protocols, version control, and human-in-loop governance. This ensures intelligence enhances operational speed without compromising regulatory confidence.
What is typical timeline to see measurable ROI?
Most organizations observe efficiency improvements within three to six months when focused on high-impact workflows such as regulatory drafting or safety case summarization. Broader enterprise gains typically follow phased expansion once pilot programs demonstrate measurable performance stability.

Nidhi Thakur
With more than 19 years of experience - I represent a team of professionals that specializes in the healthcare and business and workflow automation domains. The team consists of experienced full-stack developers supported by senior system analysts who have developed multiple bespoke applications for Healthcare, Business Automation, Retail, IOT, Ed-tech domains for startups and Enterprise Level clients.
Recent Posts

Discover how EHR, FHIR, and HL7 modernize prescription management software by improving interoperability, e-prescribing, workflow automation, and connected healthcare systems for better patient care.

Discover how an AI chatbot for healthcare improves patient engagement, automates support, integrates with EHR systems, and streamlines healthcare operations efficiently.

Modernize your medical billing workflow with automation, AI, and streamlined processes to boost efficiency, accuracy, and growth for billing companies.